
This Week In Cheminformatics: Issue #004
Random Forest Baseline Strikes Again, Better Atropisomer Prediction & Long List of Papers


Highlights
Prediction of Atropisomerism for Drug-like Molecules
In this new JCIM study, Balduf and colleagues demonstrate that standard force fields are systematically blind to this “Class 2” atropisomer, which presents barriers between 20-28 kcal/mol that allow isolation on the bench but risk racemization in vivo. Standard force fields like OPLS4 systematically overestimate rotational barriers above 30 kcal/mol. This misclassifies “Class 2” atropisomers as “Class 3” (stable enantiomers). OPLS4 correctly identified only 13 of 25 Class 2 molecules in this benchmark which is barely better than a coin flip. This suggests you cannot trust a force field to locate the transition state in all cases. Their proposed workflow uses OPLS4 solely for rough filtration, then geometry optimization and TS search is done by QRNN-TB (a semi-empirical ML potential). The results show locating the TS on the ML-corrected surface before the final DFT energy score improves accuracy drastically in particular RMSE drops from 5.4 kcal/mol (OPLS4) to 2.0 kcal/mol (QRNN-TB/DFT) and Accuracy jumps from 69% to 91%, correctly flagging 20 out of 25 Class 2 risks.
Kinetic predictions for SN2 reactions using the BERT architecture: comparison and interpretation
In this Digital Discovery study, Wilson et al. benchmark a transformer-based BERT model against a Random Forest (RF) baseline for predicting SN2 reaction kinetics, revealing that both machine learning approaches significantly outperform traditional quantum mechanics. The models achieved an RMSE of approximately 1.1 log k on external test data, surpassing high-level CCSD(T) calculations (RMSE 1.9 log k) while reducing prediction costs from hours to milliseconds. Although the RF model was vastly faster to train (256 seconds versus BERT’s 53 hours), the transformer architecture demonstrated superior chemical reasoning in specific domains, successfully learning solvent effects purely from text tokens and extrapolating reliably to reaction rates outside the training distribution. However, the study clearly highlights blind spots in the BERT model as it failed to recognize the rate-enhancing allylic effects of aromatic groups that the simpler Random Forest model correctly identified.
Long List
Cheminformatics
Uncertainty Quantification in Molecular Machine Learning for Property Predictions under Data Shifts
Context-aware Computer Vision for Chemical Reaction State Detection
Explainable active learning framework for ligand binding affinity prediction
Applications of modular co-design for de novo 3D molecule generation
A Relative Binding Free Energy Framework for Structurally Dissimilar Molecules
Universal feature selection for simultaneous interpretability of multitask datasets
A large language model-guided reinforcement learning framework for EGFR anticancer drug design
DynoPore─A Package to Analyze Molecular Dynamics Trajectories of Confined Liquids
Unified Graph-Based Interatomic Potential for Perovskite Structure Optimization
ScopeMap: An AI-Assisted, Human-in-the-Loop Workflow for Mapping Reaction Scope and Boundaries
pyEF: A Python Framework for QM and QM/MM Atom-Wise Electric Field Analysis
scII: Dual-Threshold Adaptive Integration of Single-Cell Multiomics Data Driven by Imputation
Evaluating In-Context Learning in Large Language Models for Molecular Property Regression
Assigning the Stereochemistry of Natural Products by Machine Learning
Prediction of Protein–Ligand Binding Affinities Using Atomic Surface Site Interaction Points
Multi-Solvent Graph Neural Network for Reduction Potential Prediction Across the Chemical Space
Machine-Learning Methods for pH-Dependent Aqueous-Solubility Prediction
Hemolytik 2: An Updated Database of Hemolytic Peptides and Proteins
Conformational Transition of the CARF Domain Driven by Binding Free Energy
EvoDiffMol: Evolutionary Diffusion Framework for 3D Molecular Design with Optimized Properties
Discovery of a Covalent Small-Molecule eEF1A1 Inhibitor via Structure-Based Virtual Screening
ME-pKa: A Deep Learning Method with Multimodal Learning for Protein pKa Prediction
DNACSE: Enhancing Genomic LLMs with Contrastive Learning for DNA Barcode Identification
MedChem
Reviews
Other
Polymerization mechanism of dopamine resolved: A story of strong π stacking
Click-to-Release Reactions for Tertiary Amines and Pyridines
Palate Cleanser
Breslow’s carbene and the glory of prediction
 Anish Acharya@illsciencedev reviewing their 57th PR from product manager who just discovered claude code
Anish Acharya@illsciencedev reviewing their 57th PR from product manager who just discovered claude code 3:19 AM · Jan 13, 2026 · 319K Views115 Replies · 273 Reposts · 8.38K Likes
3:19 AM · Jan 13, 2026 · 319K Views115 Replies · 273 Reposts · 8.38K Likes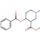 Jack of Many Spades@LoganView64206@MoleculesGoHard I always loved that joke about how you can spot a chemist in the restroom because they wash their hands *before* they use the toilet.8:16 AM · Jan 18, 2026 · 414 Views1 Repost · 9 Likes
Jack of Many Spades@LoganView64206@MoleculesGoHard I always loved that joke about how you can spot a chemist in the restroom because they wash their hands *before* they use the toilet.8:16 AM · Jan 18, 2026 · 414 Views1 Repost · 9 Likes






K bye,
Manas